
向经典看齐”是生态环境研究中心主任朱永官院士发起,由生态环境研究中心青年学术委员会委员解读经典论文的系列活动。本活动旨在鼓励青年科研人员勇于挑战高难度的科学问题,抢占科技制高点,向本领域顶尖的科学家看齐,力争取得原创性、颠覆性成果,传承经典、砥砺前行。
解读人:肖可青 林嘉铭
作者:J. Craig Venter, Karin Remington, John F. Heidelberg, Aaron L. Halpern, Doug Rusch, Jonathan A. Eisen, Dongying Wu, Ian Paulsen, Karen E. Nelson, William Nelson, Derrick E. Fouts, Samuel Levy, Anthony H. Knap, Michael W. Lomas, Ken Nealson, Owen White, Jeremy Peterson, Jeff Hoffman, Rachel Parsons, Holly Baden-Tillson, Cynthia Pfannkoch, Yu-Hui Rogers, Hamilton O. Smith
文章标题:Environmental Genome Shotgun Sequencing of the Sargasso Sea
文献来源:Science
被引次数:3,083次(Web of Science);5,653次(Google Scholar)
1.研究内容
微生物是驱动生物地球化学循环的核心,但绝大多数微生物“不可培养”的特性严重制约我们解析其生态功能。传统非培养研究依赖基于rRNA基因的PCR扩增技术探究微生物组成。然而,该技术对保守区通用引物的依赖性及引物偏好性,无法真实反映复杂环境样本中微生物群落结构与物种多样性。为突破这一技术局限,本研究通过使用全基因组鸟枪法(WGS)测序技术,揭示海水中微生物群落组成,深度挖掘未知物种与功能基因分布。
2.研究思路及核心发现
研究思路
本研究以百慕大海岸附近马尾藻海为研究区域,使用孔径为0.1–3.0 µm的过滤器富集环境微生物。通过全基因组技术,构建插入片段长度为2–6 kb的质粒基因组文库。在序列组装过程中,为避免组装器(Celera Assembler)将覆盖深度远高于平均水平的序列误判为重复序列并予以屏蔽,识别深度覆盖非冗余重叠群(contigs)。对低丰度序列,完整保留未组装的配对末端读段及单片段(singletons),以确保稀有物种信息不丢失。为应对样品复杂度高难题,研究团队将序列与非冗余氨基酸(nraa)数据库中的细菌蛋白进行比对,反向确定编码框。通过分析边界密码子,确定开放阅读框范围,进而精确识别起始与终止密码子。该方法在超过700 MB的数据集中成功鉴定出1,214,207个基因,其数量级远超当时所有已知蛋白质数据库。
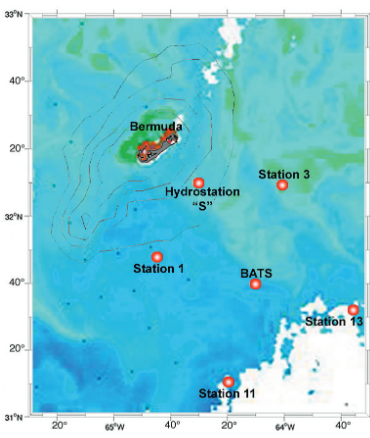
图1 2003年2月22日马尾藻海BATS站点附近海域表层叶绿素分布及采样点位。绿色色阶指示叶绿素浓度较高的区域,即高马尾藻细胞密度站位。
核心发现:
1.测序结果揭示了藻类种群变异连续性与未知性
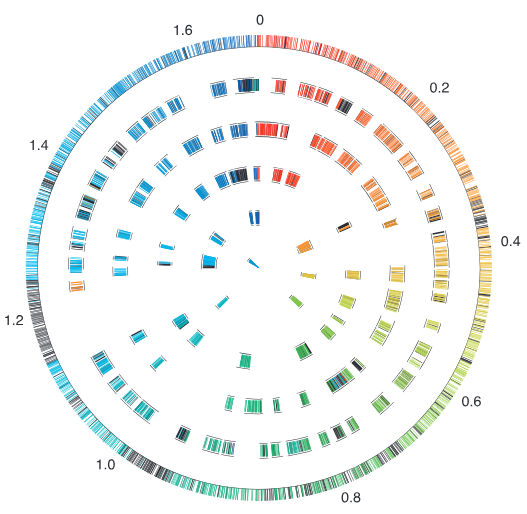
图2将环境微生物基因组与标准原绿球藻MED4基因组进行比对,结果表明超过85%的MED4基因组可与环境基因组序列匹配。内圈异色区域揭示大规模基因组重排事件;黑色区域标注表示环境样本中特有外源病毒基因簇插入。MED4发生高度功能分化,编码表面多糖合成的基因簇在环境样本中完全缺失,表明原绿球藻并非离散的单一菌株,而是高度多样化的种群连续体。
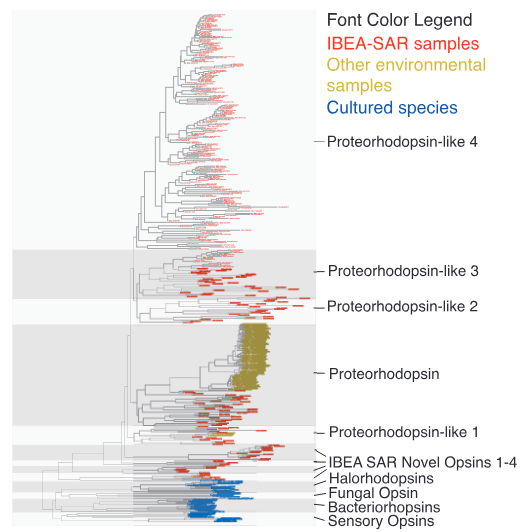
图3系统发育树展示马尾藻海样品中视紫红质(rhodopsin)类基因进化关系。蓝色为已知培养物种,黄色为其他环境来源的未培养微生物,红色为本研究在马尾藻海中新发现的未培养序列。结果表明,未培养序列在进化上远离已知培养物种,大量密集的红色分支显著扩展了现有的系统发育树。该结果证明,基于视紫红质的非叶绿素光能利用系统在海洋浮游细菌群落中广泛存在,并呈现丰富的遗传多样性。
2.比对证实了鸟枪法能够深度还原表层海水古菌谱系
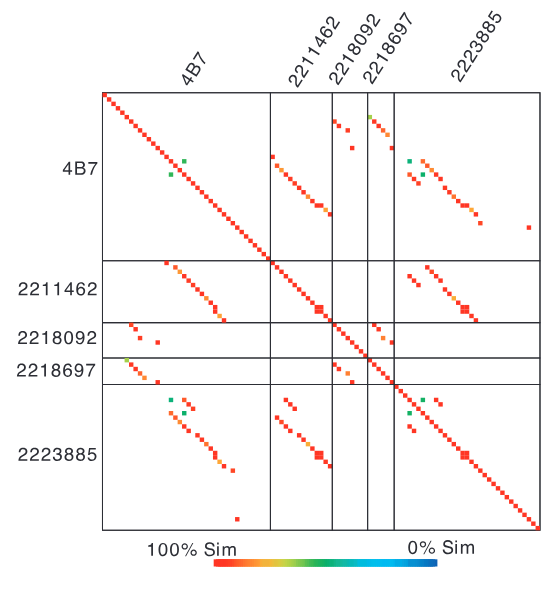
图4马尾藻海环境基因组与已知深海古菌克隆组4B7全序列比对结果。对角线轨迹表明,环境基因组与参考克隆组在基因序列上具有高度一致性。表明深海古菌谱系与海洋表层微生物共享,全基因组鸟枪法能有效解析表层海水古菌谱系组成。
3.鸟枪法较传统PCR扩增法更能准确还原微生物种群丰度
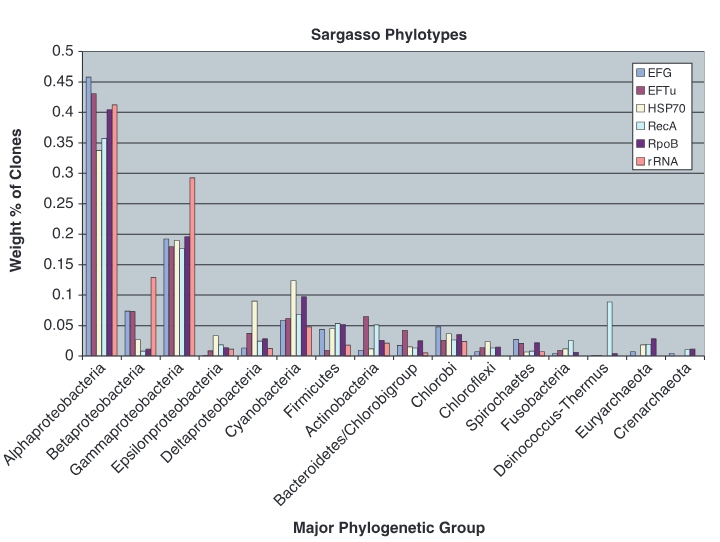
图5利用16S rRNA、RecA、EF-Tu等6种系统发育标记方法比较马尾藻海微生物群落结构。各标记物均显示变形菌门为优势类群,基于16S rRNA测序结果与单拷贝蛋白质编码基因估算生物量相对丰度偏差较大。鸟枪法避免了PCR引物偏好性导致的系统误差,能够准确反映群落中各谱系相对丰度,显著优于传统rRNA基因扩增方法。
创新突破点:
1.首次将高通量全基因组鸟枪法 (WGS) 成功应用于复杂环境样本
本研究直接对马尾藻海混合环境DNA样品进行全基因组鸟枪法测序,突破传统方法依赖PCR扩增16SrRNA基因导致的偏差,摆脱对微生物纯培养的依赖,规避PCR引物扩增盲区。
单一基因组分析中,独特区域的组装覆盖深度应近似服从泊松分布。传统组装算法在处理丰度差异巨大的混合环境样本时,易将相对丰度最高的基因序列误判为重复序列并予以屏蔽。为此,研究团队将预期覆盖深度调整为23×,识别出一组大型、深度覆盖且非冗余的重叠群(contigs)。
传统生态学研究常以rRNA基因估算物种丰富度,但不同原核生物间rRNA基因拷贝数差异显著,易导致群落比例估算出现偏差。相比之下,全基因组测序可同步获取大量rRNA序列及单拷贝蛋白编码基因,后者可作为系统发育标记物,从而准确评估群落多样性。
4.奠定了现代基因组学与新一代高通量测序的发展基石
本研究确立了“环境DNA提取—高通量测序—序列组装—分箱挖掘”研究范式,奠定现代微生物生态学中全基因组等核心生物信息学分析体系基础,预测了参与氮磷循环等元素循环新的微生物类群,其中氨氧化古菌隔年便被分离证实,开创了氮循环研究的新领域。
基因组测序策略发展概述:
1971年:引物延伸测序法由杰出华人科学家吴瑞提出使用,通过人工合成引物杂交DNA模板,引导DNA聚合酶定向合成新链,使用同位素标记法测量新链碱基种类与数量 (Wu and Taylor 1971),该方法重塑了现代生物科学发展进程。
1977年:Sanger双脱氧链终止法与引物步移法(primer walking)由F. Sanger提出使用,该时期测序采用逐段测序法,其成本高、通量低,仅覆盖病毒、质粒或单基因片段(Sanger et al. 1977),但为宏基因组技术出现奠定基础。
1995年:克隆法与鸟枪法由Venter团队提出使用。克隆法通过构建物理图谱辅助拼接,鸟枪法则直接随机打断后计算组装, 实现全基因组序列随机测序-计算组装,首次获得了较完整测序结果 (Fleischmann et al. 1995)。
2005年:二代测序法(NGS)由M. Margulies提出使用。该方法引入边合成边测序与微阵列并行技术,具有高通量测序特点 (Margulies et al. 2005)。全基因组测序成本大幅下降,推动宏基因组学、群体遗传学及临床检测的广泛应用。但短读长固有局限导致复杂重复区域存在组装缺陷。
2009年:三代测序,包括由J. Eid提出使用PacBio单分子实时测序 (Eid et al. 2009),由J. Clarke提出使用Oxford Nanopore纳米孔测序法 (Clarke et al. 2009),能够实现长读长测序目标,可直接跨越重复区与结构变异区域,充分填补着丝粒、端粒等测序盲区。
主要作者信息:

J. 克雷格·文特尔(1946-2026),鸟枪法之父,宏基因组测序技术奠基人,J. 克雷格·文特尔研究所(JCVI)创始人、董事会主席兼首席执行官。他是基因组学和合成生物学领域的先驱,其职业生涯充满了颠覆性与争议性,因此也被称为“科学狂人”。曾力排众议采用全基因组鸟枪法(WGS)与计算机算法,加速国际人类基因组计划完成,为现代医学奠定基础。同时开展全球海洋考察,利用测序技术建立全球海洋微生物基因组数据库,极大的推动了海洋微生物生态学发展。带领团队创造了全球首个由全人工合成DNA驱动的人造生命(JCVI-syn1.0),培育出仅含473个基因的最小合成细胞,带领人类实现了从读取到编写生命的历史性跨越。
参考文献:
Clarke, J., H.-C. Wu, L. Jayasinghe, A. Patel, S. Reid, and H. Bayley. 2009. Continuous base identification for single-molecule nanopore DNA sequencing. Nature Nanotechnology 4:265-270.
Eid, J., A. Fehr, J. Gray, K. Luong, J. Lyle, G. Otto, P. Peluso, D. Rank, P. Baybayan, B. Bettman, A. Bibillo, K. Bjornson, B. Chaudhuri, F. Christians, R. Cicero, S. Clark, R. Dalal, A. deWinter, J. Dixon, M. Foquet, A. Gaertner, P. Hardenbol, C. Heiner, K. Hester, D. Holden, G. Kearns, X. Kong, R. Kuse, Y. Lacroix, S. Lin, P. Lundquist, C. Ma, P. Marks, M. Maxham, D. Murphy, I. Park, T. Pham, M. Phillips, J. Roy, R. Sebra, G. Shen, J. Sorenson, A. Tomaney, K. Travers, M. Trulson, J. Vieceli, J. Wegener, D. Wu, A. Yang, D. Zaccarin, P. Zhao, F. Zhong, J. Korlach, and S. Turner. 2009. Real-Time DNA Sequencing from Single Polymerase Molecules. Science 323:133-138.
Fleischmann, R. D., M. D. Adams, O. White, R. A. Clayton, E. F. Kirkness, A. R. Kerlavage, C. J. Bult, J.-F. Tomb, B. A. Dougherty, J. M. Merrick, K. McKenney, G. Sutton, W. FitzHugh, C. Fields, J. D. Gocayne, J. Scott, R. Shirley, L.-l. Liu, A. Glodek, J. M. Kelley, J. F. Weidman, C. A. Phillips, T. Spriggs, E. Hedblom, M. D. Cotton, T. R. Utterback, M. C. Hanna, D. T. Nguyen, D. M. Saudek, R. C. Brandon, L. D. Fine, J. L. Fritchman, J. L. Fuhrmann, N. S. M. Geoghagen, C. L. Gnehm, L. A. McDonald, K. V. Small, C. M. Fraser, H. O. Smith, and J. C. Venter. 1995. Whole-Genome Random Sequencing and Assembly of Haemophilus influenzae Rd. Science 269:496-512.
Margulies, M., M. Egholm, W. E. Altman, S. Attiya, J. S. Bader, L. A. Bemben, J. Berka, M. S. Braverman, Y.-J. Chen, Z. Chen, S. B. Dewell, L. Du, J. M. Fierro, X. V. Gomes, B. C. Godwin, W. He, S. Helgesen, C. H. Ho, G. P. Irzyk, S. C. Jando, M. L. I. Alenquer, T. P. Jarvie, K. B. Jirage, J.-B. Kim, J. R. Knight, J. R. Lanza, J. H. Leamon, S. M. Lefkowitz, M. Lei, J. Li, K. L. Lohman, H. Lu, V. B. Makhijani, K. E. McDade, M. P. McKenna, E. W. Myers, E. Nickerson, J. R. Nobile, R. Plant, B. P. Puc, M. T. Ronan, G. T. Roth, G. J. Sarkis, J. F. Simons, J. W. Simpson, M. Srinivasan, K. R. Tartaro, A. Tomasz, K. A. Vogt, G. A. Volkmer, S. H. Wang, Y. Wang, M. P. Weiner, P. Yu, R. F. Begley, and J. M. Rothberg. 2005. Genome sequencing in microfabricated high-density picolitre reactors. Nature 437:376-380.
Sanger, F., S. Nicklen, and A. R. Coulson. 1977. DNA sequencing with chain-terminating inhibitors. Proceedings of the National Academy of Sciences 74:5463-5467.
Wu, R., and E. Taylor. 1971. Nucleotide sequence analysis of DNA: II. Complete nucleotide sequence of the cohesive ends of bacteriophage λ DNA. Journal of Molecular Biology 57:491-511.